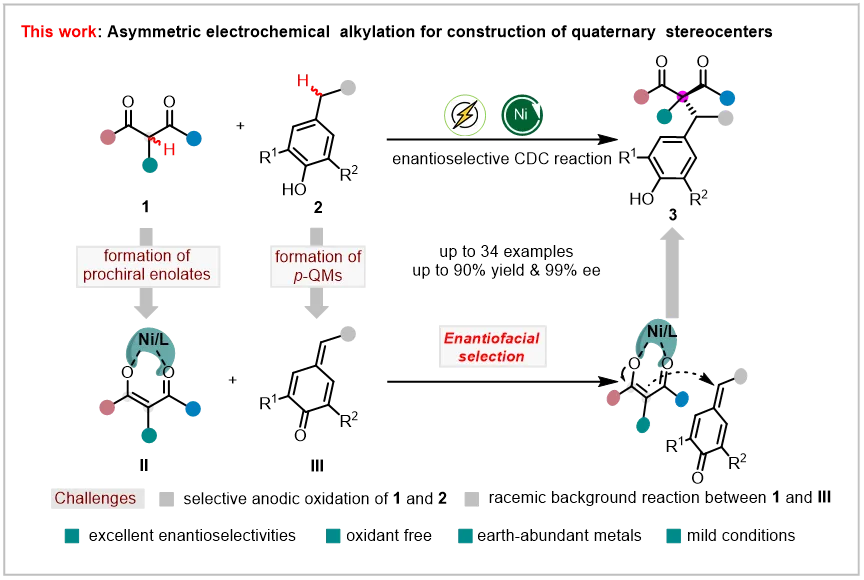
季碳手性中心广泛存在于各种药物分子、天然产物和生物活性分子中,具有结构多样性和刚性等特点。由于受位阻拥挤、构象多变等因素的影响,高效构筑季碳立体手性中心一直是有机合成中的一个巨大挑战。
含有α-H的醛、酮或酯类化合物不仅在有机合成中应用广泛且易于获得,同时也是生成前手性的C(sp2)物种的优势前体,通过后续亲电试剂(E+)对烯醇氧负离子的π面选择性进攻形成新的化学键,即可产生含有季碳手性中心的高价值化合物 (图1a)。过渡金属催化的羰基化合物α-位对映选择性官能化是一种高效构建含有季碳手性中心的羰基类化合物的策略。但是在以往的研究中,反应的底物往往需要预先官能团化,导致总的反应步骤繁琐并且原子经济性较低(图1b)。而通过C-H/C-H的转化来实现季碳手性中心的构建因更加绿色与可持续性,已被认为是有机合成中最直接的且原子经济性最高的方法之一。龚流柱课题组、冯小明课题组和刘磊课题组在这一方面做出了开创性的工作,然而上述反应往往都需要用到当量甚至过量的化学氧化剂,同时,利用C-H/C-H的不对称交叉脱氢偶联(CDC)反应来实现季碳手性中心的构建的例子依旧比较少(图1c)。

图1. 通过α位可烯醇化的羰基化合物来构筑季碳手性中心基于该研究团队对不对称电化学的研究兴趣(Angew. Chem. Int. Ed. 2024, 63, e202401355;Eur. J. Org. Chem. 2024, 27, e202400817;Chin. J. Org. Chem. 2024, 44, 748-779),徐浩等希望利用电催化与路易斯酸协同催化来实现C-H/C-H的不对称CDC反应高效构筑季碳手性中心。反应设计如下(图2),1,3-二羰基化合物1在镍催化剂的作用下形成前手性的C(sp2)物种II,酚类化合物2在电化学条件下氧化为对亚甲基苯醌(p-QMs)中间体III,接着II和III发生对映选择性地1,6-加成得到目标产物3。然而该反应仍然存在许多挑战,例如(1)1,3-二羰基化合物1自身的电氧化或者电还原可能会产生高活性的碳自由基中间体,导致其他副反应的发生;(2)电化学原位生成的p-QMs 中间体III需要克服拥挤的化学环境,才能实现季碳手性中心的构筑;(3)酚类化合物2的电氧化速率必须与前手性C(sp2)物种II的生成速率相匹配才能有效避免潜在的背景反应的发生。
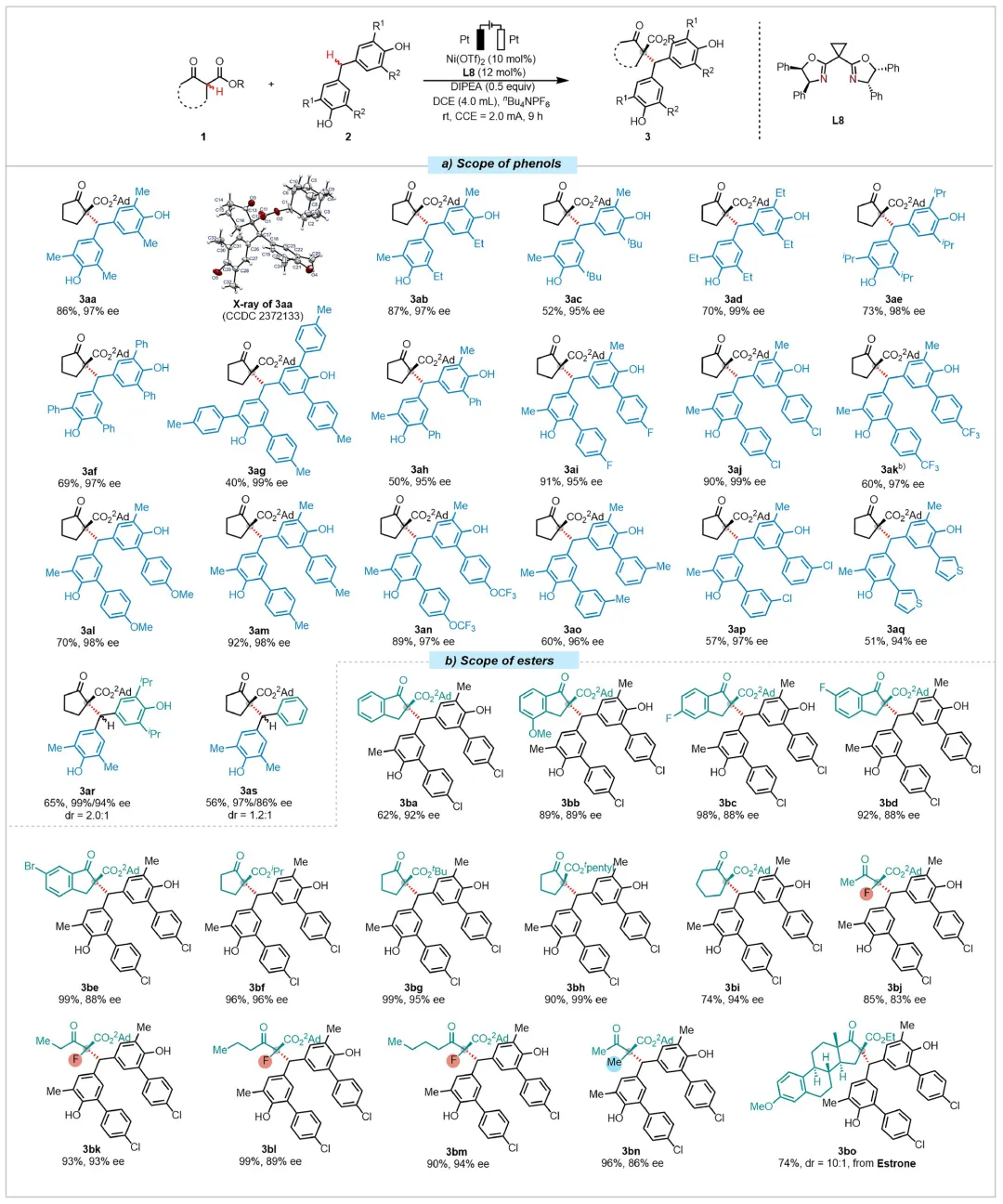
首先作者以β-酮酯1-Et和双酚类化合物2a作为标准底物,筛选了一系列手性配体(详见原文)。最终发现当使用手性BOX配体L8时,可以以79%的产率,64%的ee值得到目标产物3a。接着作者在此基础上,筛选了不同的路易斯酸、碱等条件,均不能提高反应的对映选择性。令作者惊喜的是,当使用异丙基β-酮酯1-iPr时,反应的对映选择性大幅提升(表1,entry 8)。接着作者进一步增大酯基部分的位阻,使用2-金刚烷基β-酮酯1a时,反应最高能以86%的产率,97%的ee值得到目标产物3aa。接着作者又进行了一系列控制实验及与传统氧化剂的对比实验,证明了在该反应中电流及路易斯酸催化剂的重要性(详见原文)。
 a) Reaction conditions: undivided cell, Pt anode (1.5*1.0 cm2), Pt cathode (1.5*1.0 cm2), 1-Et (0.45 mmol, 1.5 equiv, R = Et), 2a (0.3 mmol, 1.0 equiv), Lewis acid (0.03 mmol, 10 mol%), L8(0.036 mmol, 12 mol%), Cp2Fe (0.03 mmol, 10 mol%), Base (0.15 mmol, 0.5 equiv), nBu4NPF6 (0.3 mmol, 1.0 equiv), DCE (4.0 mL), constant current = 2.0 mA, 9 h, rt. Isolated yield. Enantiomeric excess of 3a, 3b, 3aa were determined by HPLC analysis using a chiral stationary phase. b) Substrate 1-iPr was used, R = iPr. c) Substrate 1a was used, R = 2-adamantyl. d) Entry 9 without Cp2Fe. e)Entry 10 without ligand. f) Entry 10 without electricity. g) 1H NMR yield based on 1,1,2,2-tetrachloroethane as the internal standard.
a) Reaction conditions: undivided cell, Pt anode (1.5*1.0 cm2), Pt cathode (1.5*1.0 cm2), 1-Et (0.45 mmol, 1.5 equiv, R = Et), 2a (0.3 mmol, 1.0 equiv), Lewis acid (0.03 mmol, 10 mol%), L8(0.036 mmol, 12 mol%), Cp2Fe (0.03 mmol, 10 mol%), Base (0.15 mmol, 0.5 equiv), nBu4NPF6 (0.3 mmol, 1.0 equiv), DCE (4.0 mL), constant current = 2.0 mA, 9 h, rt. Isolated yield. Enantiomeric excess of 3a, 3b, 3aa were determined by HPLC analysis using a chiral stationary phase. b) Substrate 1-iPr was used, R = iPr. c) Substrate 1a was used, R = 2-adamantyl. d) Entry 9 without Cp2Fe. e)Entry 10 without ligand. f) Entry 10 without electricity. g) 1H NMR yield based on 1,1,2,2-tetrachloroethane as the internal standard.
在确定了最佳反应条件之后,作者对底物普适性范围进行了考察(图3)。首先是一系列邻位烷基取代的双酚类衍生物均能兼容于该反应,以较高的产率和对映选择性得到目标产物(3aa-3ae)。更难氧化的邻位芳基取代的双酚类衍生物也能以优异的对映选择性得到目标产物(3af-3ag)。当R1基团固定为甲基时,R2基团可以兼容各种取代的芳基或者杂芳基,以中等至优秀的产率和优异的对映选择性得到目标产物(3ah-3aq)。作者还尝试了亚甲基处取代基不同的酚类衍生物,其也能以适中的产率和优异的对映选择性得到目标产物(3ar-3as),但是dr值较低。之后作者对一系列β-酮酯类化合物进行了底物拓展。一系列不同芳基取代的β-酮酯类化合物均能成功兼容于该反应(3ba-3be)。值得注意的是,β-酮酯的酯基也不仅仅局限于2-金刚烷基,更为普适的异丙基、叔丁基、特戊基也均能兼容于该反应,以优异的对映选择性得到目标产物(3bf-3bh)。同时,六元环、开链以及复杂药物分子衍生的β-酮酯类化合物也能成功兼容于该反应(3bi-3bo),由此可以实现更具挑战的开链含F季碳手性中心的构筑。
季碳手性中心广泛存在于各种药物分子、天然产物和生物活性分子中,具有结构多样性和刚性等特点。由于受位阻拥挤、构象多变等因素的影响,高效构筑季碳立体手性中心一直是有机合成中的一个巨大挑战。
含有α-H的醛、酮或酯类化合物不仅在有机合成中应用广泛且易于获得,同时也是生成前手性的C(sp2)物种的优势前体,通过后续亲电试剂(E+)对烯醇氧负离子的π面选择性进攻形成新的化学键,即可产生含有季碳手性中心的高价值化合物 (图1a)。过渡金属催化的羰基化合物α-位对映选择性官能化是一种高效构建含有季碳手性中心的羰基类化合物的策略。但是在以往的研究中,反应的底物往往需要预先官能团化,导致总的反应步骤繁琐并且原子经济性较低(图1b)。而通过C-H/C-H的转化来实现季碳手性中心的构建因更加绿色与可持续性,已被认为是有机合成中最直接的且原子经济性最高的方法之一。龚流柱课题组、冯小明课题组和刘磊课题组在这一方面做出了开创性的工作,然而上述反应往往都需要用到当量甚至过量的化学氧化剂,同时,利用C-H/C-H的不对称交叉脱氢偶联(CDC)反应来实现季碳手性中心的构建的例子依旧比较少(图1c)。
图1. 通过α位可烯醇化的羰基化合物来构筑季碳手性中心
基于该研究团队对不对称电化学的研究兴趣(Angew. Chem. Int. Ed. 2024, 63, e202401355;Eur. J. Org. Chem. 2024, 27, e202400817;Chin. J. Org. Chem. 2024, 44, 748-779),徐浩等希望利用电催化与路易斯酸协同催化来实现C-H/C-H的不对称CDC反应高效构筑季碳手性中心。反应设计如下(图2),1,3-二羰基化合物1在镍催化剂的作用下形成前手性的C(sp2)物种II,酚类化合物2在电化学条件下氧化为对亚甲基苯醌(p-QMs)中间体III,接着II和III发生对映选择性地1,6-加成得到目标产物3。然而该反应仍然存在许多挑战,例如(1)1,3-二羰基化合物1自身的电氧化或者电还原可能会产生高活性的碳自由基中间体,导致其他副反应的发生;(2)电化学原位生成的p-QMs 中间体III需要克服拥挤的化学环境,才能实现季碳手性中心的构筑;(3)酚类化合物2的电氧化速率必须与前手性C(sp2)物种II的生成速率相匹配才能有效避免潜在的背景反应的发生。
首先作者以β-酮酯1-Et和双酚类化合物2a作为标准底物,筛选了一系列手性配体(详见原文)。最终发现当使用手性BOX配体L8时,可以以79%的产率,64%的ee值得到目标产物3a。接着作者在此基础上,筛选了不同的路易斯酸、碱等条件,均不能提高反应的对映选择性。令作者惊喜的是,当使用异丙基β-酮酯1-iPr时,反应的对映选择性大幅提升(表1,entry 8)。接着作者进一步增大酯基部分的位阻,使用2-金刚烷基β-酮酯1a时,反应最高能以86%的产率,97%的ee值得到目标产物3aa。接着作者又进行了一系列控制实验及与传统氧化剂的对比实验,证明了在该反应中电流及路易斯酸催化剂的重要性(详见原文)。
a) Reaction conditions: undivided cell, Pt anode (1.5*1.0 cm2), Pt cathode (1.5*1.0 cm2), 1-Et (0.45 mmol, 1.5 equiv, R = Et), 2a (0.3 mmol, 1.0 equiv), Lewis acid (0.03 mmol, 10 mol%), L8(0.036 mmol, 12 mol%), Cp2Fe (0.03 mmol, 10 mol%), Base (0.15 mmol, 0.5 equiv), nBu4NPF6 (0.3 mmol, 1.0 equiv), DCE (4.0 mL), constant current = 2.0 mA, 9 h, rt. Isolated yield. Enantiomeric excess of 3a, 3b, 3aa were determined by HPLC analysis using a chiral stationary phase. b) Substrate 1-iPr was used, R = iPr. c) Substrate 1a was used, R = 2-adamantyl. d) Entry 9 without Cp2Fe. e)Entry 10 without ligand. f) Entry 10 without electricity. g) 1H NMR yield based on 1,1,2,2-tetrachloroethane as the internal standard.
在确定了最佳反应条件之后,作者对底物普适性范围进行了考察(图3)。首先是一系列邻位烷基取代的双酚类衍生物均能兼容于该反应,以较高的产率和对映选择性得到目标产物(3aa-3ae)。更难氧化的邻位芳基取代的双酚类衍生物也能以优异的对映选择性得到目标产物(3af-3ag)。当R1基团固定为甲基时,R2基团可以兼容各种取代的芳基或者杂芳基,以中等至优秀的产率和优异的对映选择性得到目标产物(3ah-3aq)。作者还尝试了亚甲基处取代基不同的酚类衍生物,其也能以适中的产率和优异的对映选择性得到目标产物(3ar-3as),但是dr值较低。之后作者对一系列β-酮酯类化合物进行了底物拓展。一系列不同芳基取代的β-酮酯类化合物均能成功兼容于该反应(3ba-3be)。值得注意的是,β-酮酯的酯基也不仅仅局限于2-金刚烷基,更为普适的异丙基、叔丁基、特戊基也均能兼容于该反应,以优异的对映选择性得到目标产物(3bf-3bh)。同时,六元环、开链以及复杂药物分子衍生的β-酮酯类化合物也能成功兼容于该反应(3bi-3bo),由此可以实现更具挑战的开链含F季碳手性中心的构筑。
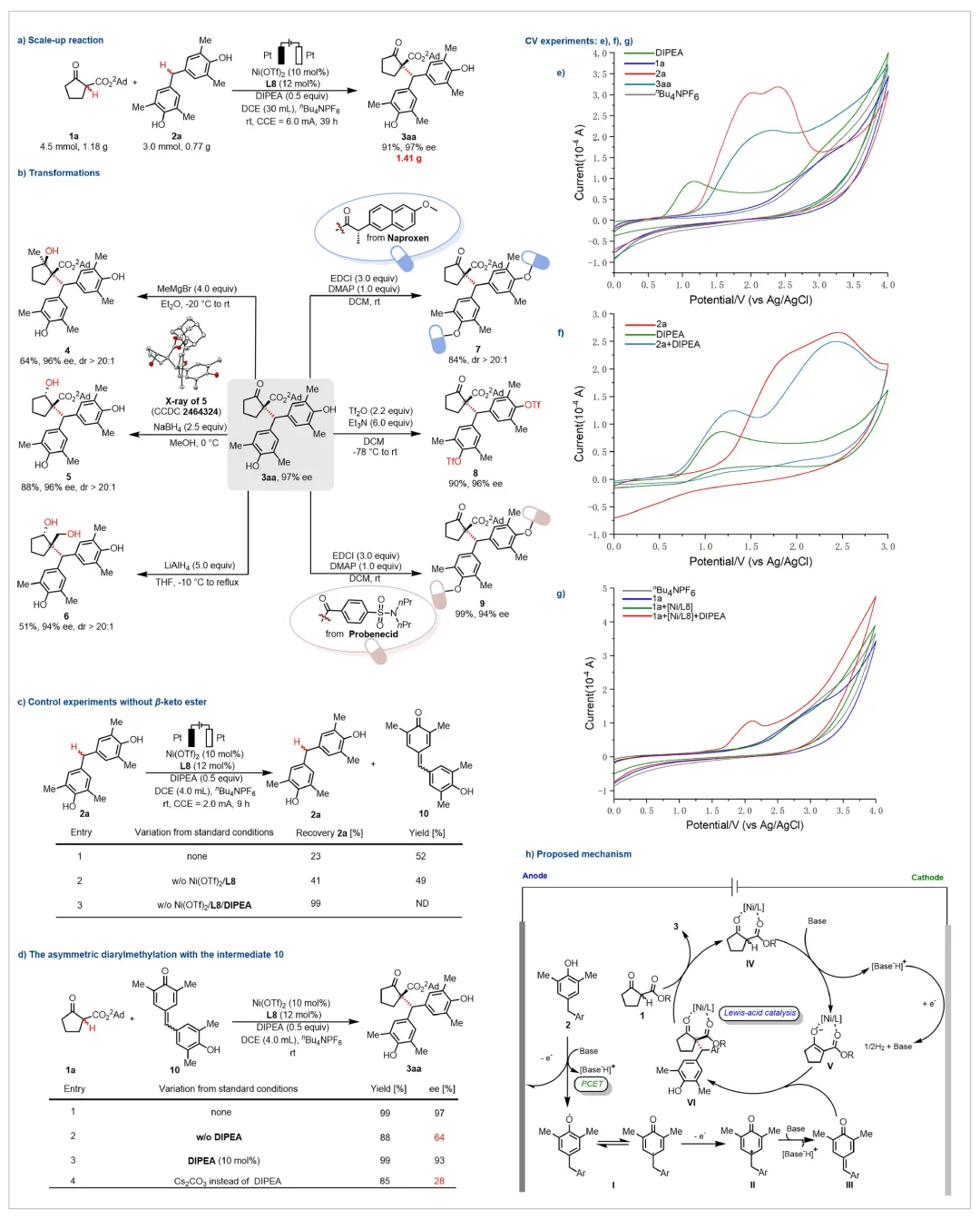
为了进一步证明了该反应的实用性,作者进行了克量级规模实验,当将反应底物的量扩大10倍时,该反应依然能以91%的产率和97%的ee值得到目标产物(图4a)。同时所得到的产物3aa也可以进行丰富的转化。产物3aa中的酮羰基可以被甲基格氏试剂进攻或者硼氢化钠还原,以优异的对映选择性和非对映选择性构建手性叔醇和仲醇官能团。更为重要的是,产物3aa中金刚烷取代的酯基也可以在加热条件下被氢化锂铝还原,以优异的非对映选择性生成手性1,3二醇。而产物3aa双酚结构中的羟基则可以作为平台分子,通过一步简单的酯化反应即可对药物分子进行后修饰。同时,酚羟基还可以进一步转化为OTf基团,从而使得产物可以通过C-O键的断裂实现更为丰富的转化(图4b)。
之后作者对反应机理进行了探究,为了验证反应是否产生了设想的p-QM中间体10,作者进行了控制实验(图4c),当在标准条件下单独电氧化双酚类衍生物2a时,作者能以52%的分离产率得到p-QM 10。这说明p-QM 10极有可能是反应的关键中间体。后续的控制实验表明,所使用的碱DIPEA在p-QM 10的生成过程中起到至关重要的作用。之后,作者提前制备了p-QM 10, 在不通电的条件下与β-酮酯1a进行反应,该反应能以高达99%的产率和97%的ee值得到目标产物,这证明了p-QM 10是反应的关键中间体(图4d)。后续的控制实验表明,所使用的碱DIPEA在控制反应的对映选择性方面也起到了关键作用。作者也进行了循环伏安实验来进一步验证反应机理(图4e-4g)。作者发现,当在双酚衍生物2a中加入DIPEA时,2a的第一个氧化峰的氧化电势会大幅下降,从1.98 V降低至1.27 V,根据实验现象以及文献的报道,作者推测DIPEA的加入,可能使得2a可以在较低的氧化电位下通过PCET过程发生氧化生成p-QM 中间体10。通过CV图(图4g),作者发现只有当在β-酮酯1a和Ni催化剂中加入DIPEA的时候,CV图中才能出现一个明显的氧化峰(E = 2.10 V vs. Ag/AgCl),作者推测该峰是由更易发生单电子氧化的烯醇类化合物V产生的。因此,作者认为DIPEA的加入促进了前手性的C(sp2)物种V的生成,其更好的手性环境有利于反应中对映选择性的控制。
基于以上机理验证实验,作者提出反应可能的机理:在碱的作用下,双酚衍生物2经历质子耦合电子转移(PCET)过程以产生酚氧自由基物种I。随后I的阳极氧化和去质子化产生关键中间体p-QM III。同时,β-酮酯1与手性镍催化剂配位得到中间体IV。中间体IV在碱的作用下生成前手性的C(sp2)物种V,V随后与p-QM III进行选择性地1,6-加成反应,得到相应的中间体VI。接着中间体VI将镍催化剂释放到未反应的β-酮酯1中,得到目标产物3.
综上所述,作者开发了一种新型的镍催化电化学不对称CDC反应,用于构建羰基α位季碳手性中心。该高效且可持续的方法,避免了氧化剂和贵金属催化剂的使用,在温和条件下以中等至优秀的产率和优异的对映选择性实现了一系列含有羰基α位季碳手性中心化合物的构建。该类型的电催化不对称CDC反应在驱动开发新型的不对称CDC反应中具有重要意义。
相关成果近期在线发表于Science China Chemistry。博士研究生钱伟烽为论文的第一作者,朱翠菊副教授和徐浩教授为论文的共同通讯作者。在本研究工作DFT计算过程中,武汉大学戚孝天教授与华中师范大学张之涵教授提出了诸多建设性意见与宝贵建议,作者谨此致以诚挚感谢。详细内容见:Nickel-catalyzed electrochemical asymmetric diarylmethylation for construction of quaternary carbon stereocenters. Wei-Feng Qian, Yan-Yan Ouyang, Tong Liu, Ting Fang, Zhi-Yong Fan, Jin-Lin Zhang, Cuiju Zhu*, Hao Xu*. Sci. China Chem. 2025, DOI:10.1007/s11426-025-2919-y.


