近期,华中师范大学化学学院张国柱/郭瑞研究团队在光诱导铜催化领域取得多项研究进展,相关成果相继发表在J. Am. Chem. Soc.,Angew. Chem. Int. Ed.,ACS Catal.,Green Chem.等化学领域顶级期刊上。
一、配体进化驱动的对映选择性C(sp3)−H叠氮化反应

光学纯脂肪族有机叠氮化物被认为是有机合成中的多功能合成子,能够进行广泛的转化,包括Curtius重排、Staudinger反应、Huisgen“点击”反应等。尽管在近几十年中付出了巨大努力并取得了显著进展,但将叠氮基团催化不对称地引入有机分子中仍然相对罕见。迫切需要一种通用方法,能够高效且对映选择性地在复杂分子中引入叠氮基团。显然,以高化学选择性、区域选择性和立体选择性为特征的直接C(sp³)−H键叠氮化是一种更具吸引力且成本效益更高的策略,有可能显著缩短合成路线,并实现复杂分子的高效后期修饰(Scheme 1a)。
羰基化合物是众多天然产物和功能分子的基本构建模块。它们在化学和物理性质上的固有稳定性,加上它们的高生物相容性,使其在药物化学中不可或缺。手性α-叠氮羰基化合物作为广泛生物活性分子和药物中的核心单元发挥着关键作用(Scheme 1b,右)。此外,它们代表了极其重要且具有战略意义的功能前体,可以转化为多种重要的光学纯化合物,如非天然氨基酸、氨基醇和含氮杂环(Scheme 1b,左)。因此,开发新的活化策略并筛选出高效手性配体以实现α-羰基C−H键的不对称叠氮化是特别引人注目的研究领域。
课题组报道一种新策略实现了复杂羰基化合物的不对称α-C(sp³)−H叠氮化。使用TMSN3作为叠氮源,利用光诱导铜催化的芳基自由基介导的分子内1,5-HAT策略,反应在温和条件下顺利进行,生成具有高对映选择性(高达98%ee)的脂肪族叠氮化物。这一化学的成功归功于结合手性配体修饰和配体自组装的配体进化策略(Scheme 1c,ii)。值得注意的是,这一方法表现出显著的官能团兼容性,能够在多种生物活性天然产物、药物和复杂分子中引入叠氮基团(Scheme 1c,iii)。此外,所得的手性α-叠氮羰基产物易于进行后续转化,便于合成各种高价值的非天然氨基酸衍生物。
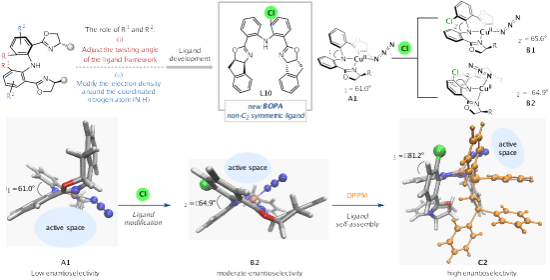
课题组在铜催化的不对称自由基交叉偶联反应取得成功的基础上,进一步测试了一系列N,N,N-三齿阴离子手性双恶唑啉二苯胺(BOPA)配体。作者探索了在苯环上引入各种取代基以微调电子效应和立体效应(Scheme 2)。计划从两个关键方面对手性BOPA配体进行修饰:(i)通过在二苯胺的苯环邻位引入立体位阻不同的取代基,旨在调整配体框架的扭曲角度,微妙地改变手性环境;(ii)引入电子性质不同的取代基将改变配位氮原子(N−H)周围的电子密度,进而影响高价铜中间体的稳定性。这些方法旨在提高最终C−N键形成步骤的效率,并进一步调控立体选择性。同时,双(二苯基膦)甲烷(DPPM)作为添加剂时,能够显著提升产物的立体选择性。DFT研究表明,DPPM的存在构建了一个“立体墙”,进一步增大了扭曲角度(−81.2°),并压缩了活性空间,从而为C(sp³)−H叠氮化反应提供了高对映选择性(Scheme 2)。
Scheme 1. Background and Concept Design 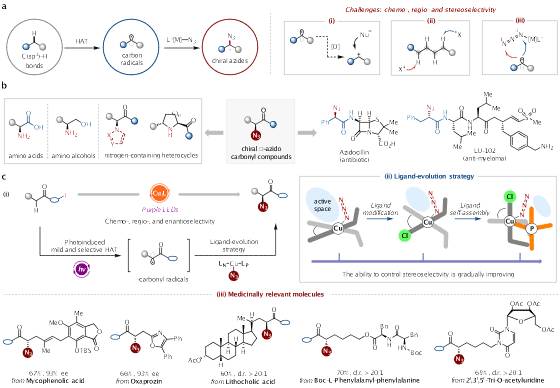
Scheme 2. Ligand evolution
Scheme 3. Substrate scope for enantioselective α-C(sp3)-H azidation of amides
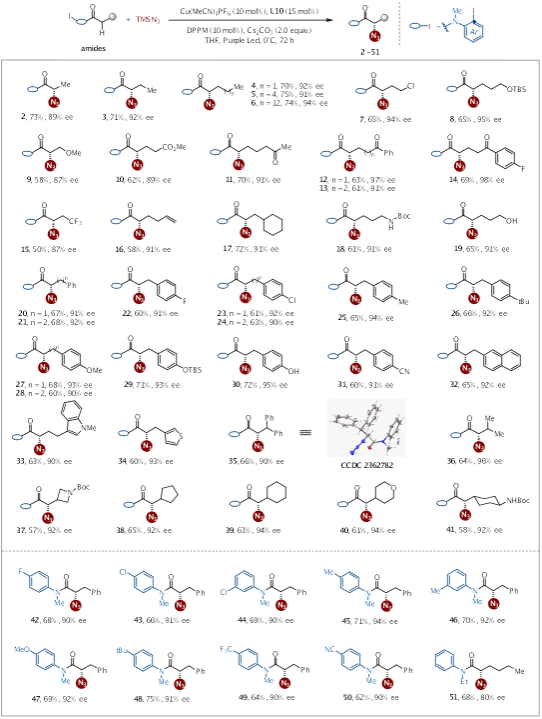
在最优条件下,作者探索了不对称C(sp³)−H叠氮化反应的普适性。对于多种常见官能团,如Cl、OTBS、OMe、CO2Me、酮、CF3、烯烃和环己基,均能很好地耐受,以高对映选择性得到目标产物。该体系对于含有取代基的苯丙酰胺底物同样适用。这种方法也适用于萘基和杂芳基底物,生成所需的光学活性叠氮化物。对于β位带有取代基的酰胺,包括二苯基、二甲基和不同环大小的环烷基基团的酰胺,如高度应变的氮杂环丁烷,在温和条件下耐受良好,生成高ee值的产物。值得注意的是,环烷基基团上的杂原子和取代基的存在并不影响反应的对映选择性。
作者对含有药物分子片段、天然产物衍生的酰胺进行修饰,以中等到良好产率和优异区域选择性、对映选择性和非对映选择性(d.r. > 20:1)得到手性叠氮化产物。这些例子表明,不对称C(sp³)−H叠氮化策略适用于复杂生物活性分子的后期修饰,并且能够耐受一系列杂环和官能团。此外,这种方法的多功能性突显了其在生物活性测试和药物开发中进一步应用的潜力。
Scheme 4. Late-stage modification of natural products and medicinally relevant molecules

Scheme 5. Mechanistic Investigation 
作者通过机理实验验证了反应过程。KIE实验证明分子内1,5-HAT不是该叠氮化反应的速率限制步骤(Scheme 5a)。自由基捕获实验证明反应是通过自由基途径进行的(Scheme 5c)。EPR实验证明了叠氮自由基的存在(Scheme 5d)。HRMS实验证实了BOPA-Cu(I)和BOPA-Cu(I)-DPPM复合物的存在(Scheme 5e),而X射线晶体学确认了BOPA-Cu(II)复合物81的结构(Scheme 5f)。EPR实验也验证了BOPA-DPPM-Cu(II)-N3中间体的存在(Scheme 5g)。荧光猝灭实验表明,只有激发态Cu(I)-BOPA复合物,而不是BOPA-Cu(I)-DPPM复合物,能被酰胺S1有效猝灭,证实了BOPA-Cu(I)复合物在该反应中作为主要光催化剂(Scheme 5h)。配体L10的ee与产物1的ee之间的近乎线性相关性表明,铜中心可能只与一个手性BOPA配体配位(Scheme 5i)。此外,开/关光照射实验和量子产率(Φ = 0.005%)排除了自由基链机制的可能性。综上作者提出了可能机理:在碱存在下,BOPA-Cu(I)复合物I与TMSN3反应生成BOPA-Cu(I)-N3复合物II。在紫光LED照射下,激发态的Cu(I)复合物III通过单电子转移(SET)过程与芳基碘化物IV反应,生成BOPA-Cu(II)-N3复合物V和芳基自由基VI。随后,芳基自由基VI通过分子内1,5-氢原子转移(HAT)过程转化为关键的α-羰基自由基物种VII。与此同时,关键的手性铜催化剂BOPA-DPPM-Cu(II)-N3(VIII)可能通过两种不同的路径生成:在途径A中,复合物V通过光诱导的LMCT过程生成叠氮自由基并再生BOPA-Cu(I)复合物I,从而完成光催化循环。随后,叠氮自由基被BOPA-Cu(I)-DPPM复合物IX捕获,生成BOPA-DPPM-Cu(II)-N3复合物VIII。在途径B中,DPPM与V的配位可逆地生成复合物VIII。最终,所需的光学活性叠氮化产物通过α-羰基自由基物种VII与BOPA-DPPM-Cu(II)-N3复合物VIII的自由基交叉偶联反应生成,同时再生复合物IX并完成手性催化循环。在这其中,DPPM从IX中的解离也可再生BOPA-Cu(I)复合物I。
综上所述,课题组开发了一种高度对映选择性的α-C(sp³)−H叠氮化反应,适用于复杂羰基化合物,使用TMSN3作为叠氮源。通过利用光诱导铜催化的芳基自由基介导的分子内1,5-HAT策略,该反应在温和条件下顺利进行,生成具有卓越化学选择性、区域选择性和对映选择性的手性脂肪族有机叠氮化物。这一化学反应的成功归功于结合手性配体修饰和配体自组装的配体进化策略。这一方法表现出显著的官能团兼容性,能够在多种生物活性天然产物、药物和复杂分子中引入叠氮基团。此外,生成的手性α-叠氮羰基产物易于进行后续转化,便于合成高价值的非天然氨基酸衍生物,并且可以作为点击化学的连接平台,直接将叠氮基团引入复杂生物活性化合物中。因此,该方法的多功能性突显了其在生物活性测试和药物开发中进一步应用的潜力。机理研究与DFT计算结果表明,C−N键的形成是通过α-羰基自由基与铜(II)上的叠氮基团之间的远程SH2机制实现的,而高对映选择性则由与铜(II)相连的叠氮基团以及DPPM的苯环构建的紧凑活性手性空间所保证。相关研究成果发表在J. Am. Chem. Soc.上。博士研究生张赫和古维为该论文的第一作者,计算部分由武汉大学戚孝天教授团队完成。(DOI:10.1021/jacs.5c16924)
二、光氧化还原催化与三重氢原子转移策略实现酰胺与非活化烯烃的氢烷基化
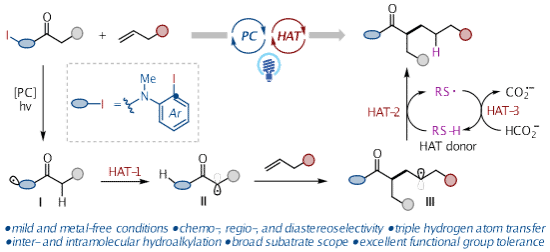
该反应在温和无金属条件下实现了分子间与分子内的氢烷基化,展现出优异的化学、区域和非对映选择性,同时具备广泛的底物适用性和出色的官能团耐受性。包括乙烯在内的多种短链气态烯烃也能成功参与反应。通过结构复杂生物活性分子的多样化后期修饰与合成,进一步凸显了该方法的通用性。机理研究表明,该反应通过包含分子内与分子间步骤的三重氢原子转移(HAT)过程进行,其成功实施得益于弱亲电性α-羰基自由基的可控生成,以及自由基极性匹配/错配效应的策略性应用,从而精准调控不同的碳自由基物种与未活化烯烃或HAT供体间的反应活性。这一结合了光氧化还原催化与多重氢原子转移的策略,在解决惰性C(sp³)–H键活化及非活化烯烃氢烷基化等长期挑战性问题方面具有重要潜力。相关研究成果发表在Angew. Chem. Int. Ed.上。文章在审稿过程中得到审稿人的一致高度评价(Top 5%), 并被编辑选为VIP论文。硕士研究生易贝贝为该论文的第一作者。(Angew. Chem. Int. Ed. 2025, e202516794. DOI:10.1002/anie.202516794)
三、光诱导铜催化芳基自由基介导1,5-HAT实现胺的α-C(sp3)−H键叠氮化
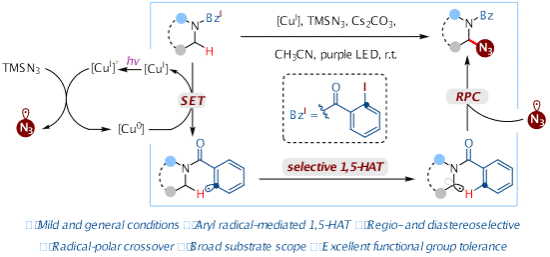
课题组以2-碘苯甲酰基团作为自由基迁移基团,发展了一种光诱导铜催化的原料胺与TMSN3的α-叠氮化反应。该方法具有条件温和、通用且氧化还原中性的特点,具备广泛的底物适用性、优异的官能团耐受性以及高区域选择性和立体选择性,可实现多种复杂生物活性分子的后期叠氮化。所得α-叠氮胺可方便地转化为各类含氮杂环及其他官能团,该策略将在合成化学与药物发现领域展现重要应用价值。相关研究成果发表在ACS Catal.上。博士研究生张赫和古维为该论文的共同第一作者。计算部分由武汉大学戚孝天教授团队完成。(ACS Catal. 2025, 15, 15425−15434. DOI:10.1021/acscatal.5c04218)
四、借助极性匹配的氢原子转移策略实现光诱导铜催化烯烃的对映选择性烷基/炔基化
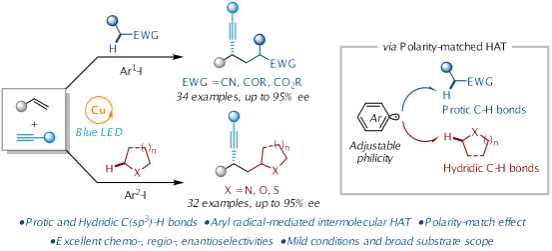
课题组开发了一种光诱导铜催化的对映选择性三组分烷基炔基化反应,通过极性匹配的氢原子转移(HAT)策略,实现了从广泛可用的化学原料直接合成手性炔烃。该方法不仅首次实现了以简单乙腈为底物的对映选择性自由基交叉偶联反应,还扩展到了其他含有质子C(sp³)−H键的化合物,如酮、酯、酰胺、醚和胺,展现了优异的化学、区域和立体选择性。这一成果不仅为合成手性分子提供了一种高效、绿色的途径,还为未来设计更多基于烷基C(sp³)−H键活化的不对称自由基交叉偶联反应提供了重要的理论和实验基础。相关研究成果发表在J. Am. Chem. Soc.上。硕士研究生吴政泽和李凯为该论文的共同第一作者。(J. Am. Chem. Soc. 2025, 147, 22072−22083. DOI:10.1021/jacs.5c06054)
五、光诱导铜催化区域和立体选择性多组分[3+2+1]自由基环化反应合成四氢吡啶
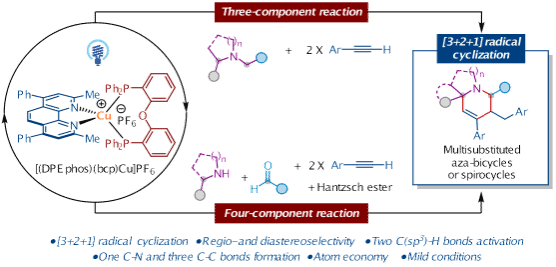
课题组开发了一种光诱导铜催化的三组分或四组分[3+2+1]自由基环化反应,从容易获得的原料胺、炔烃和醛出发,制备传统方法较难合成的多取代的双环或螺环四氢吡啶。该方法条件温和、原子经济性高,具有优异的区域选择性和立体选择性,并且能够在单步反应中同时活化两个α-氨基C(sp³)-H键,形成三个C-C键和一个C-N键。相关研究成果发表在Org. Lett.上。博士研究生李思佳和李建烨为该论文的共同第一作者。(Org. Lett. 2025, 27, 5057−5062. DOI:10.1021/acs.orglett.5c00865)
六、光诱导铜催化环氧化物和炔烃的自由基阴离子交叉偶联反应
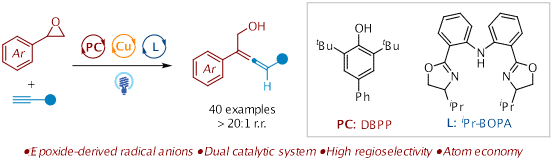
课题组开发了一种温和的光诱导铜催化的自由基交叉偶联反应,用于环氧化物和末端炔烃的反应,能够以优异的区域选择性合成多取代的联烯醇。该双催化体系能够耐受广泛的芳基、杂芳基和烷基炔烃,多样化的环氧化物底物以及复杂的天然产物衍生物,并以中等到良好的收率提供具有合成价值的联烯醇骨架。该双催化体系避免了使用化学计量的添加剂,不仅扩展了环氧化物功能化的工具箱,还强调了在温和、可持续条件下现代有机合成中自由基阴离子化学的实用性。相关研究成果发表在Green Chem.上。博士研究生杨晨为该论文的第一作者。(DOI:10.1039/d5gc03812j)
七、铜催化非活化烯烃的对映选择性三组分氟烷基/炔基化反应
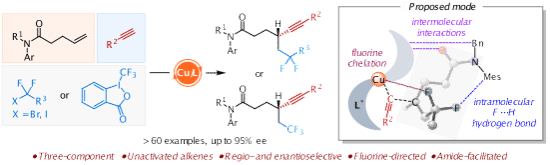
课题组首次开发了一种铜催化的非活化烯烃的高区域选择性和对映选择性氟烷基炔基化反应,使用一系列末端炔烃和多样的氟烷基卤化物在温和条件下进行。除了氟烷基卤化物,Togni试剂也可以参与反应,生成具有高对映选择性的手性β-三氟甲基炔烃。该方法展示了优异的官能团耐受性和广泛的底物范围,能够对多种生物活性化合物进行后期修饰。使用大位阻的茚基取代的BOPA配体是该化学反应成功的关键。通过开发更多的金属催化体系、手性配体以及多样的电亲和亲核试剂,我们预计这种螯合辅助策略将在非活化烯烃的不对称三组分双官能化中展现出巨大的潜力,能够构建更具挑战性的手性碳−碳和碳−杂原子键。相关研究成果发表在ACS Catal.上。博士研究生廖梦霞为该论文的第一作者。计算部分由武汉大学戚孝天教授团队完成。(ACS Catal. 2025, 15, 1693−1703. DOI:10.1021/acscatal.4c06641)
八、光诱导铜催化通过1,5-氢原子转移策略实现酰胺的多样性转化
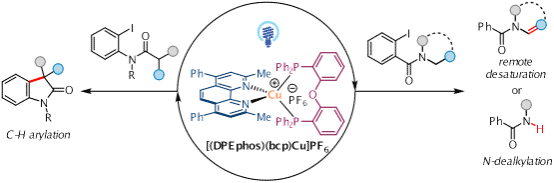
课题组开发了一种通过光诱导铜催化的分子内1,5-氢原子转移策略实现酰胺多样性转化的方法。这种方法能够将酰胺底物转化为三种不同的产物:C-H芳基化、远程脱饱和和N-脱烷基化。一系列的开链和环状胺以及生物相关的杂环胺在温和条件下均具有良好的兼容性。这种策略为合成异吲哚和烯胺化合物提供了一种更具成本效益和环境友好的方法。相关研究成果发表在ChemCatChem上。博士研究生潘春香和硕士研究生张梦珍为该论文的第一作者。(ChemCatChem 2025, 17, e202402146. DOI:10.1002/cctc.202402146)
以上工作得到国家自然科学基金(22301092、22271112 和 22201222)、中国中央高校基础研究基金(CCNU24JCPT016)、中国国家重点研发计划(2022YFA1505100 和 2023YFA1508600)、武汉英才人才计划、华中师范大学(CCNU)的启动资金以及武汉大学超算中心的超级计算系统提供的财政支持。